Molecular Creator: A Quantum-Inspired Approach to Molecular Design and Simulation
Abstract: In this article, we present the development of a quantum-inspired Molecular Creator project within the BSM-SG Engine, integrating charge interactions, electron orbitals, and atomic structure based on BSM-SG theory. This innovative simulation tool allows for the precise visualization and interaction with molecular structures by leveraging CIF (Crystallographic Information File) loading, enabling the simulation and prediction of new materials and pharmaceuticals. Our approach is designed to bridge quantum physics, computational chemistry, and real-world applications, offering a unique solution for drug discovery and material design.
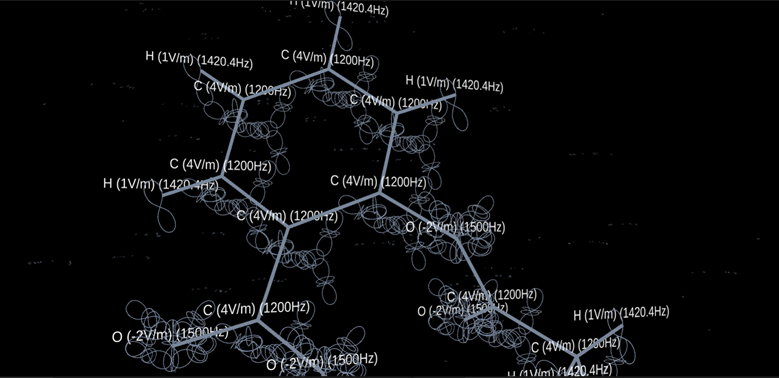
[Fig. 1 Aspirin Molecule]
1. Introduction
Understanding molecular structures and their interactions is a fundamental challenge in chemistry, physics, and material sciences. Classical computational tools often rely on approximations that do not fully capture the quantum nature of atomic interactions. Our Molecular Creator project integrates BSM-SG theoretical principles to simulate atomic-level charge distributions, proton-neutron interactions, and electron orbital configurations, providing an interactive and highly precise molecular modeling platform.
By utilizing BSM-SG Engine, the system allows real-time visualization and manipulation of molecular structures, making it accessible to both researchers and developers. A key feature of this project is the ability to import and process CIF files, enabling the simulation of existing and hypothetical molecular configurations.
2. Theoretical Framework
2.1 The BSM-SG Atomic Model
Unlike conventional quantum mechanical models that rely on probabilistic electron clouds, BSM-SG (Basic Structures of Matter – Supergravitational) theory introduces geometrically defined orbits for electrons, structured around intrinsic charge distributions. This model enables us to predict molecular stability, charge interactions, and bonding with higher precision.
Key features of the BSM-SG atomic model include:
- Defined electron orbits instead of probability-based wavefunctions.
- Stationary protons within atomic nuclei with dynamic neutron-induced magnetic stabilization.
- Electrostatic and magnetic charge interactions that dictate molecular formations.
- Quantum-level charge balance influencing molecular binding and lattice configurations.
3. Methodology
3.1 Molecular Visualization
The simulation is built using real-time rendering capabilities, allowing for dynamic manipulation and visualization of molecules. Molecular data is extracted from CIF files, which contain crystallographic information, including:
- Atomic positions
- Bonding angles
- Lattice structures
3.2 Charge Interactions and Electron Orbitals
Each atom is represented as a charge system, where:
- Protons define the central charge potential.
- Neutrons contribute to the magnetic moment, stabilizing the nucleus.
- Electrons orbit within quantized bands influenced by neighboring atoms.
The system applies Coulombic force equations and magnetostatic interactions to predict molecular stability and reaction dynamics.
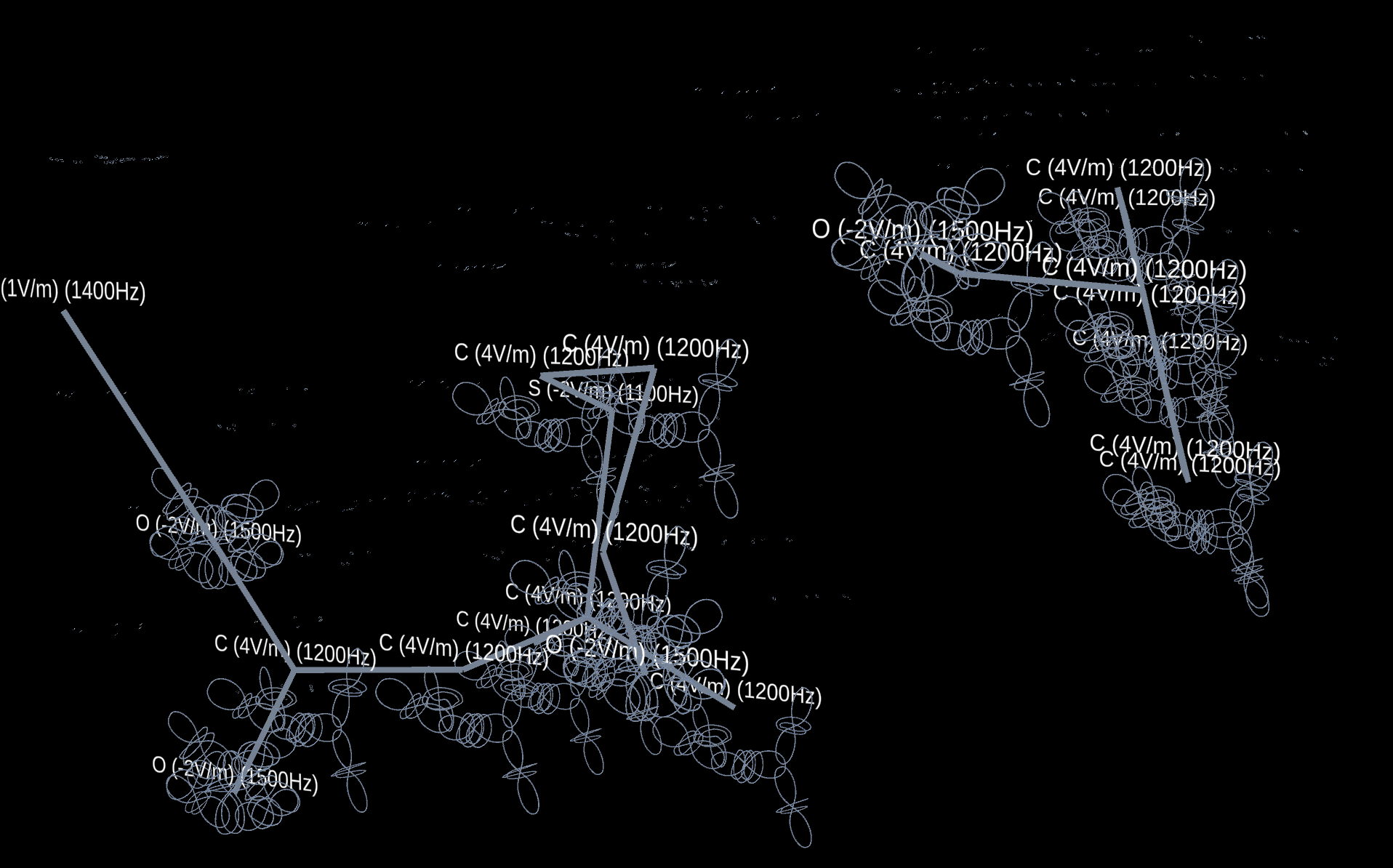
[Fig.2 Penicillin Molecule]
4. Applications
4.1 Drug Discovery
The ability to simulate molecular interactions provides a platform for drug design, allowing researchers to test molecular binding in a virtual environment before laboratory synthesis.
4.2 Material Science and Nanotechnology
The project enables researchers to design new composite materials by analyzing molecular formations and predicting the behavior of nanostructured compounds under various conditions.
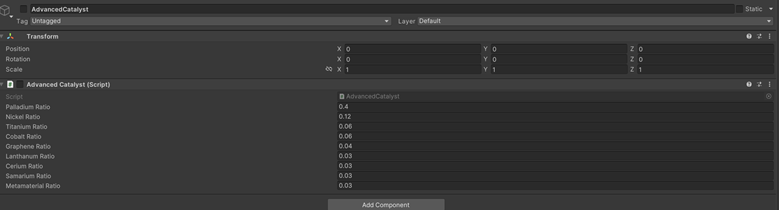
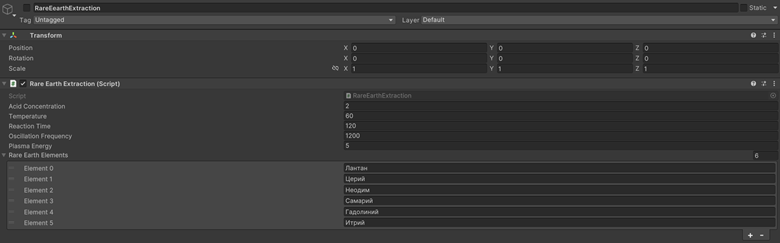
4.3 Quantum Computing and Molecular Encoding
Using the BSM-SG approach, we explore how molecules can be encoded as quantum bits (qubits) for future quantum computing applications.
5. Conclusion and Future Work
The Molecular Creator represents a new paradigm in molecular modeling, offering a quantum-accurate, visually interactive, and scalable approach to molecular simulations. By integrating BSM-SG atomic theory and charge-based interactions, this project opens new possibilities in drug discovery, materials research, and computational chemistry.
Work in progress includes:
- Expanding CIF file compatibility to include dynamic molecular simulations.
- Integrating machine learning algorithms for predictive molecular modeling.
- Connecting the platform to quantum computing frameworks for next-generation computational chemistry.
This project stands as a testament to the power of quantum-inspired computation in bridging fundamental physics and applied sciences, paving the way for revolutionary advancements in material and pharmaceutical research.
2025 Victor Pronchev